|
PROGNOS Input Form Tutorial
Follow this tutorial to see how to input your nuclease into PROGNOS, interpret the output, and order PCR primers for experimental validation.
- For this tutorial, start by pressing the "Show Example Values" button on the PROGNOS tool page:
- PROGNOS has ranking algorithms for both Zinc Finger Nucleases (ZFN) and TAL Effector Nucleases (TALEN). For this tutorial, select "TALEN".

- TALENs and ZFNs bind to DNA in different orientations.
You can choose if you would like to enter the target of your nucleases as it appears on the top/positive DNA strand or in the locations that the nucleases bind.
- When entering the target sequence of your nucleases, you can include ambiguous letter codes.
In that case, if a potential off-target site contains any of the possible bases, it will not be counted as a mismatch.
- For example, since the "NN" RVD for TALENs is known to bind to A with relatively equal affinity as G, the code "R" could be used in place of G.
This would allow for either an A or a G to appear in that position in a potential off-target site without being counted as a mismatch.
- For TALENs, make sure to include the 5' base in your target sequence (usually a "T") in addition to the bases targeted by the RVDs.
For this tutorial, the DNA target should already appear.

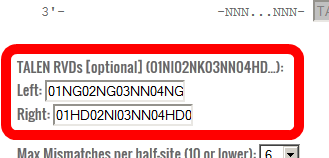
- If you are using PROGNOS on a TALEN, you may choose to enter the set of RVDs you are using. This is used for the RVDs ranking algorithm.
If this is left blank, PROGNOS will assume the standard code: NI-->A, HD-->C, NN-->G, NG-->T
These RVDs should be entered with a two digit number (01, 02...10, 11...) followed by the single letter amino acid code for the RVD. There should be no spaces.
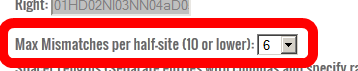
- This parameter allows you to change the level of homology to the intended target site that is required in the potential off-target sites.
In general, 3 maxmimum mismatches per half-site is a good choice for ZFNs and 6 for TALENs.
However, if you are using ZFNs or TALENs much shorter or much longer than usual, you may need to adjust this number so that the search will find a helpful number of sites.
See step #14 for more details. For this tutorial, set the maximum number of mismatches in each half-site to 6.
- Different nuclease architectures permit different allowed distances (spacings) between the two nuclease binding sites.
In general, spacers of 5 or 6 bp work well for ZFNs while TALENs cleave over a range of 10-30 bp spacings.
Different TALEN architectures have different preferences though; Figure 6 of Christian et al. describes this well.
For this tutorial, set the allowed spacer lengths to "10-30" (as short as 10 bp and as long as 30 bp).

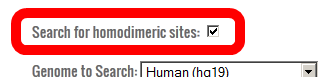
- Although TALENs and ZFNs are designed to target a sequence as heterodimers (one left and one right), the wild-type FokI endonuclease domain can homodimerize.
If you are using the standard wild-type FokI domain, you should include homodimeric sites (left-left and right-right) in your search.
If you are using obligate heterodimeric FokI domains (such as ELD/KKR, EL/KR, etc.), you can choose to only search for heterodimeric sites.
However, cleavage at homodimeric sites by hetero-obligate FokI domains has been observed in rare cases by Gabriel et al.
For this tutorial, check the box to allow homodimeric sites.
- The PROGNOS server contains many different genomes that can be searched for potential off-target sites.
If there is an additional publically available genome that you would like to see added you can express your interest in the Discussion thread.
For this tutorial, select the Human genome.

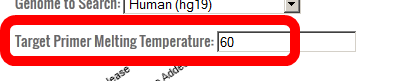
- PROGNOS will automatically design PCR primers to amplify potential off-target sites from genomic DNA.
The default target melting temperature for these primers is 60°C.
If you have a special application that requires a significantly different PCR process, you may choose to alter this parameter.
However, PCR success rates for temperatures outside of 56-63°C have not been tested and using 60°C has been validated by hundreds of successful automated designs.
For this tutorial, enter 60.
- PROGNOS will automatically design PCR primers to amplify potential off-target sites from genomic DNA.
For different applications, it is beneficial to have the nuclease site in different positions relative to the ends of the amplicon.
For example, a Surveyor Assay requires the visualization of 3 distinct bands on a gel: the uncleaved band, and two cleavage bands.
On the other hand, SMRT sequencing does not require the sequences on either side of the nuclease site to be of different lengths.
Settings optimized for use only with SMRT Sequencing or only with the Surveyor Assay are available as well as settings offering a compromise that can be used for both methods.
For different sequencing platforms or assays, custom lengths can be chosen.
For this tutorial, enter 125 for the minimum separation between uncleaved and cleaved products (which is also the distance from the edge of the amplicon to the nuclease site).
Also enter 75 for separation between cleavage bands.

- PROGNOS will list all sites in a genome that match the search criteria, but due to processing constraints, it will only rank a certain number of these sites.
Whatever number is entered here, that will be the number of top sites that PROGNOS will rank and design primers for.
Depending on how many sites you plan to experimentally validate, you can change this number, but looking at the top 48 sites will give a good overview of the off-target potential.
For this tutorial, enter 48.

- The parameter specifies how much genomic sequence surrounding the nuclease target site will be saved with your output.
PROGNOS uses this only to design primers and it may need more or less depending on the primer specifications you gave it.
Since more sequence will mean a larger file size, it is recommended to use the minimum amount necessary, which can be found by pressing the "Calculate" button.
If you have need of even more sequence for a later analysis you plan to do, you can increase the amount of extra sequence stored in the output file.
For this tutorial, enter 422.

- You can, but are not required to, enter your email address so that you will be notified when your PROGNOS results are ready.
If you do not choose to enter your email address, you should bookmark your output page so that you don't lose track of it.
For this tutorial, you can leave it blank.
- Searches can return a different number of off-target sites depending on the parameters chosen.
If the parameters you choose are too strict (i.e. only allowing 1 mismatch per half-site for a TALEN), there may be no sites in the genome with that level of homology.
If the parameters are too lax, you would receive an overwhelmingly high number of off-target sites which would overload the server.
To ensure that neither of these things happens, a calculation is made based on your input parameters to determine the number of sites expected to be found in the genome.
If this number is to high or to low, you should adjust the number of mismatches you allow per half-site. See step #5.
For this tutorial, 6 mismatches per half-site should result in ~1600 expected sites.



- Our server runs as fast as it can, but sometimes there may be other searches waiting to be performed.
This gives an estimate of when your results should be ready. Oftentimes it is much sooner, this is the maximum estimate.

- For this tutorial, do NOT press the "Submit Form" button, but this is what you would press to start your PROGNOS search running.
To continue on to learn how to interpret the results of the PROGNOS search, move on to the PROGNOS Output tutorial.
|




